 Image credit: Chao Hou
Image credit: Chao HouAbstract
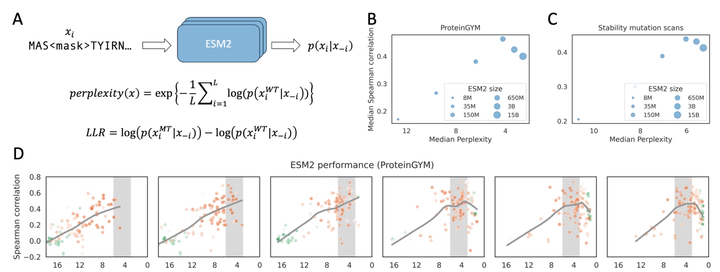
Protein language models, and models that incorporate structure or homologous sequences, estimate sequence likelihoods p(sequence) that reflect the protein fitness landscape and are commonly used in mutation effect prediction and protein design. It is widely believed in deep learning field that larger model performs better across tasks. However, for fitness prediction, language model performance declines beyond a certain size, raising concerns about their scalability. Here, we showed that model size, training dataset, and stochastic elements can bias the predicted p(sequence) away from real fitness. Model performance on fitness prediction depends on how well p(sequence) matches evolutionary patterns in homologs, which is best achieved at a moderate p(sequence) level for most proteins. At extreme predicted wild-type sequence likelihoods, models predict uniformly low or high likelihoods for nearly all mutations, failing to reflect the real fitness landscape. Notably, larger models tend to predict proteins with higher p(sequence), which may exceed the moderate range and thus reduce performance. Our findings clarify the scaling behavior of protein models on fitness prediction and provide practical guidelines for their application and future development.
Supplementary notes can be added here, including code, math, and images.